где
D
e
—
энергия диссоциации молекулы
,
D
e
=
ω
2
e
4
ω
e
x
e
;
(2)
q
=
r
−
r
e
,
r
—
межъядерное расстояние
,
r
e
—
равновесное межъядер
-
ное расстояние
;
α
=
µ
ω
e
x
e
B
e
r
2
e
¶
1
/
2
;
ω
e
,
ω
e
x
e
,
B
e
—
колебательные и вращательная постоянные
.
Известных к настоящему времени экспериментальных данных для
основных и возбужденных состояний
CuAg
и
CuAu
недостаточно для
построения потенциальных кривых РКР в широкой области измене
-
ния межъядерного расстояния
,
поэтому для аппроксимации потенциа
-
лов рассматриваемых молекул использована функция Морзе
.
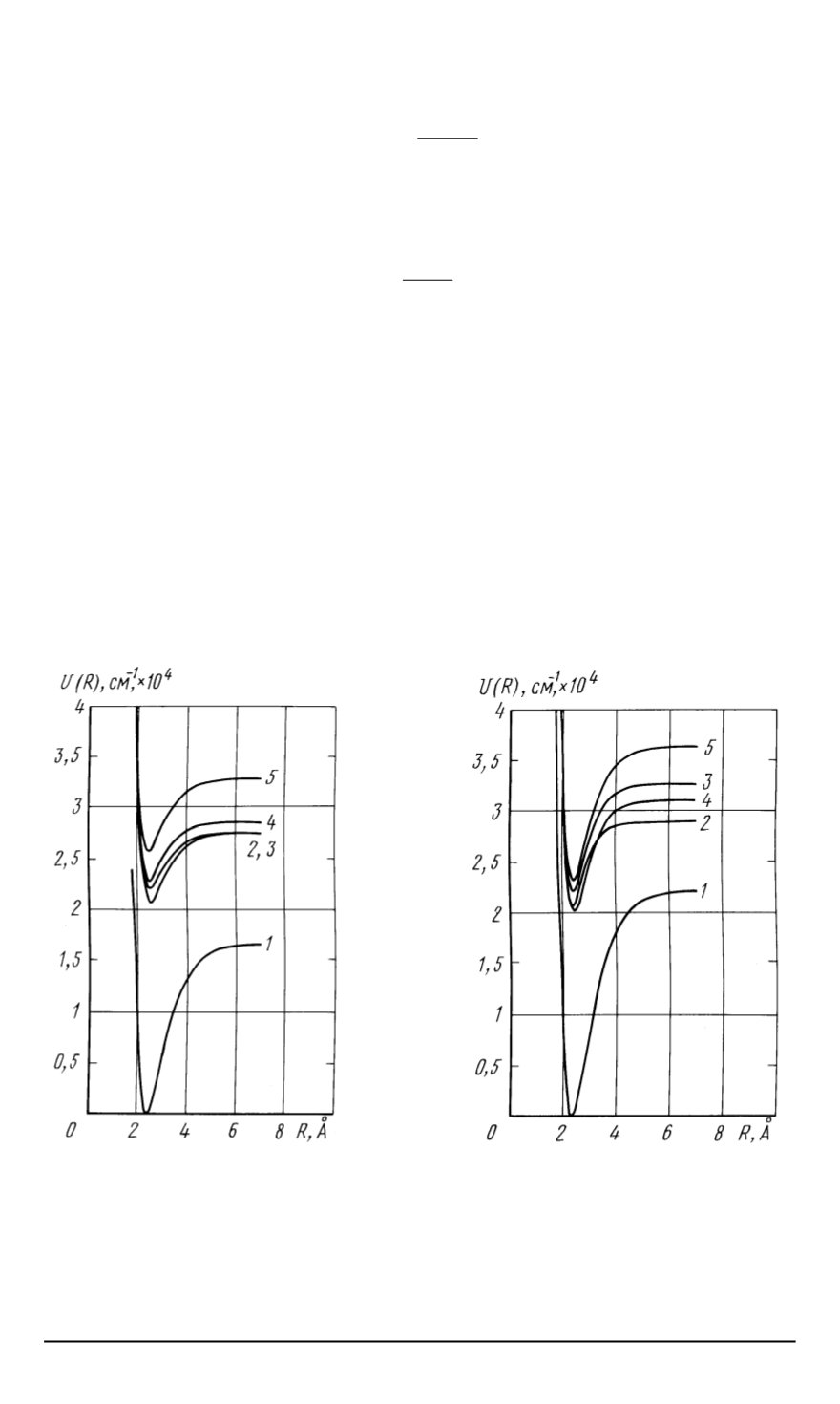
На рис
. 1,
2
приведены построенные в настоящей работе потенциальные кривые
для основных и возбужденных электронных состояний
CuAg
и
CuAu.
Расчет молекулярных постоянных проведен методом возмущений для
модели вращающегося осциллятора двухатомной молекулы
.
Рис
. 1.
Потенциальные функ
-
ции Морзе для основного и воз
-
бужденных электронных состо
-
яний молекулы
CuAg:
1
— X
1
Σ
+
,
2
— A0
+
,
3
— A1,
4
— B
1
0
+
,
5
— B0
+
Рис
. 2.
Потенциальные функции
Морзе для основного и возбужден
-
ных электронных состояний моле
-
кулы
CuAu:
1
— X
1
Σ
+
,
2
— A
0
1,
3
— B0
+
,
4
— C1,
5
— D1
74 ISSN 0236-3933.
Вестник МГТУ им
.
Н
.
Э
.
Баумана
.
Сер
. "
Естественные науки
". 2004.
№
1


